Von Rebecca R.: 2017 DNO Nationals, 2rd Place Winner
Ich durfte vom 22. bis zum 24. Mai ein sehr kurzes aber auch überaus spannendes Praktikum im Leibniz Institut für Altersforschung in Jena absolvieren. Nach den üblichen Sicherheitshinweisen am Montagmorgen konnte ich direkt am Laboralltag teilnehmen und die Präparation eines Gehirns eines Mäuse-Embryos beobachten. Die daraus extrahierten Neuronen wurden für die weitere Arbeit im Labor benötigt. Für mich war es wirklich interessant bei der Präparation, die ein hohes Maß an Präzession erforderte, zuzusehen, aber auf eine genauere Beschreibung soll an dieser Stelle verzichtet werden.
Daraufhin folgte ein spannender Vortrag einer Studentin der Arbeitsgruppe über das Thema ihrer Bachelorarbeit, welche sich mit der Erforschung heditärer spastischer Paraplegien auf zellulärer Ebene befasst.
Zum Abschluss meines ersten Tages wurde mir mein Hauptprojekt für das Praktikum erklärt, welches darin bestand, Neuronen und sogenannte HeLa-Zellen zu färben und zu mikroskopieren. HeLa-Zellen wurden ursprünglich 1951 dem Zervixkarzinom einer Patientin (Henrietta Lacks, daher die Abkürzung HeLa) entnommen und dann weiter erforscht. Dabei gelang es eine Krebszelle mit einem veränderten Teilungsverhalten (viel schnellere Teilung und kein Absterben auch nach vielen Teilungen) zu identifizieren und zu vermehren. Somit entstand eine gewissermaßen unsterbliche Zelllinie, die heute in Laboren weltweit verbreitet ist. Der erste Schritt zur Färbung der HeLa-Zellen war GFP mittels „Lipofectamine“ zu transfizieren, wodurch das Endoplasmatische Retikulum eine grüne Färbung erhielt. Dazu musste zuerst eine Lösung bestehend aus DNA, Lipofectamine und destillierten Wasser hergestellt werden. Dafür wurden jeweils 120 μL einer DNA- bzw. einer Lipofectamine-Lösung erstellt, wobei die Masse des Lipofectamines der doppelten Menge an DNA entsprach. Beide wurden daraufhin miteinander vermischt und für 5 min inkubiert. Schließlich wurde diese Mixtur zu den HeLa-Zellen hinzugefügt, welche über Nacht in einem Brutschrank inkubiert wurden. Die gesamte Prozedur musste möglichst steril erfolgen (da hierbei noch mit lebendigen Zellen gearbeitet wurde), weshalb sie unter einer Sicherheitswerkbank durchgeführt wurde.
Am nächsten Morgen durfte ich erneut einen typischen Bestandteil des Laboralltages erleben. Zuerst hörte ich mir im Rahmen eines Labortreffens dreier Arbeitsgruppen zwei Vorträge (über das Thema einer Bachelor- und Masterarbeit) an. Diese Treffen finden zweiwöchig statt, während in den übrigen Wochen kleinere Besprechungen nur innerhalb der jeweiligen Arbeitsgruppen abgehalten werden. Zudem hatte ich das Glück, dass an diesem Tag ebenfalls ein Institutstreffen stattfand, bei welchem Mitglieder aus allen Abteilungen des Instituts zusammenkommen. Während diesem wurden noch drei weitere spannende Vorträge von Doktoranden über deren Dissertationen gehalten.
Daraufhin setzte ich die Färbung der Neuronen und HeLa-Zellen fort, wobei sich die Zellen bereits auf Deckgläschen befanden, die in Zellkulturmedium kultiviert wurden. Um bestimmte Zellbestandteile zu markieren, wurde eine Immunofluoreszenz-Färbung benutzt. Dabei werden zwei verschiedene Antikörper verwendet: Ein primärer Antikörper bindet an eine für die jeweilige Komponente der Zelle spezifische Struktur, während sich der sekundäre Antikörper (welcher einen fluoreszierenden Farbstoff trägt) an den primären Antikörper anheftet. Durch die Benutzung zweier Antikörper wird die Spezifität der Bindung somit wesentlich erhöht. Doch bevor ich die Antikörper zu den Zellen hinzufügen konnte, musste ich diese zunächst dafür vorbereiten. Dazu wurden die Zellen zunächst mit Methanol (mit einer Temperatur von -20 °C) fixiert. Danach wurden Ammoniumchlorid (um Aldehyde, die ein falsch-positives Ergebnis bedingen können, zu binden) sowie 0,2 %-iges Triton-X-100 (um Membranlipide auszuwaschen) hinzufügt, wobei diese Schritte immer wieder durch das Waschen mit PBS (phosphate buffered saline) unterbrochen wurden. Zuletzt wurden unspezifische Bindungstellen für Proteine, an welche sich die Antikörper anlagern könnten, innerhalb der Zellen mithilfe eines Blockmediums beseitigt.
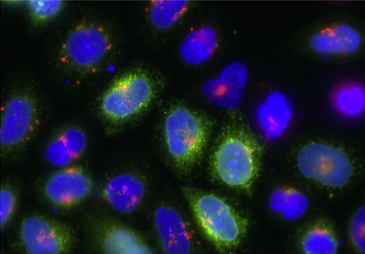
Danach konnte mit dem eigentlichen Färbevorgang begonnen werden. Dafür wurde sogenannter Parafilm verwendet, auf welchen 20 μL des jeweiligen primären Antikörpers getropft wurden. Die Deckgläschen wurden daraufhin mit den Zellen nach unten auf die Tropfen gelegt. Dabei wurde für jedes Deckgläschen ein anderer primärer Antikörper verwendet (also auf jedem wurde eine andere Struktur gefärbt). Bei den HeLa-Zellen lag das Endoplasmatische Retikulum aufgrund der Transfektion bereits gefärbt vor. Zusätzlich dazu wurden noch der Golgi-Apparat (das Protein Giantin), die Mikrotubuli (das Protein α-Tubulin) und frühe Endosomen (das Protein EEA 1 – Early Endosome Antigen 1) markiert. Dagegen wurden bei den Neuronen für diese typische Strukturen also Dendriten (das Protein MAP2), Axone (Tau-Proteine) und Axonhügel (das Protein Ankyrin G) gefärbt. Nach einer Inkubationszeit von 20 Minuten und dem zweifachen Waschen mit PBS wurden die Deckgläschen mit dem sekundären Antikörper (bei gleicher Prozedur wie bei dem primären Antikörper) für 20 Minuten jedoch unter Lichtausschluss inkubiert. Darauf folgte ein erneutes Waschen sowie die Färbung des Zellkerns bei allen Proben mittels des Farbstoffes „hoechst“ bei fünfminütiger Inkubationszeit. Zuletzt wurden die Deckgläschen mithilfe von Mowiol auf den Objektträgern fixiert, wobei das Mowiol über Nacht trocknen konnte.
Am nächsten Morgen war der Moment der Wahrheit gekommen: Endlich konnte an einem Fluoreszenzmikroskop überprüft werden, ob die Transfektion und die Färbung erfolgreich waren. Das Ergebnis waren wirklich faszinierende wunderschöne Bilder!

Es waren sehr interessante und erlebnisreiche drei Tage. Vielen Dank an die Arbeitsgruppe von Dr. Kaether – im besonderen an Laura Behrendt und Jana Hamann – dass ich so herzlich aufgenommen und intensiv betreut wurde!